
PREDICCIÓN
FUNCIONAL Y MODELADO DE REDES MOLECULARES


Dr. Juan Antonio García Ranea Predoc. Ian Morilla
ranea@uma.es ian.morilla@uma.es
Contacto:
Departamento
de Biología Molecular y Bioquímica.
Facultad
de Ciencias – Universidad de Málaga.
Campus
de Teatinos s/n 29071 Málaga – Spain.
+34
952 13 20 25.
Trayectoria Investigadora del IP:
Desde
2008:
Investigador Ramón y Cajal. Universidad de Málaga.
2002-2007: Investigador
Senior en la University College London.
2001: Doctor en
Ciencias. Universidad de Málaga.
2000: Master en
Administración y Dirección de Empresas: Executive-MBA, ICAI-ICADE, Universidad
Pontificia de Comillas. Madrid.
1996-2002: Becario de
investigación en el Centro Nacional de Biotecnología, Consejo Superior de
Investigaciones Científicas. Madrid.
1995-1996: Becario de
investigación en el Centro de Biología Molecular Severo Ochoa, Consejo Superior
de Investigaciones Científicas. Madrid.
1992-1995: Estudiante
interno y posteriormente becario de investigación en el Departamento de
Genética, Universidad de Málaga.
1994: Licenciado en
Ciencias Biológicas. Universidad de Málaga.
Líneas de
Investigación:
1.
Predicción,
modelado y análisis topológico de redes de proteínas para los proteomas
completos de H.
sapiens y Saccharomyces
cerevisiae.
En estrecha colaboración
con el grupo de la profesora Christine Orengo en la University College London
(UCL), hemos modelado dos tipos de redes de interacción entre proteínas para
los proteomas completos de H. sapiens y S. cerevisiae, redes que denominamos KnowledgeGram (KG) y PredictoGram
(PG) por el origen de los datos usados en su construcción. Las redes KG
integran la información sobre asociación funcional de pares de proteínas
obtenida de bases de datos de “conocimiento” biológico y/o experimental, tales
como: HRPD, MINT, Intact, Reactome, Kegg, Gene Ontology (GO) y FunCat. Mientras
que las redes PG, integran mediante el método Fisher, predicciones ab-initio de
interacción entre proteínas obtenidas por tres métodos bioinformáticos
distintos diseñados y desarrollados en éste mismo trabajo, como son: GECO (Gene
Expressison COrrelation). CODA (Co-Ocurrence Domain Analysis); y hiPPI
(homology inherited Protein-Protein Interactions).
En este trabajo
demostramos que las redes PG, basadas en predicciones, presentan las mismas
características topológicas de las redes biológicas experimentales KG. También
demostramos que las redes PG contienen información funcional clave en el
modelado de los sistemas biológicos que componen estos dos organismos
estudiados, información que en un alto porcentaje (80%-90%) no está recogida en
las redes KG basadas en evidencias experimentales.
2.
Modelado y
análisis evolutivo de los complejos de proteínas en los proteomas completos de Escherichia coli
y Saccharomyces
cerevisiae.
En colaboración con la UCL
hemos modelado con alta precisión los complejos de proteínas para los proteomas
completos de E.
coli y S.
cerevisiae, basándonos en complejos de proteínas comúnmente aceptados por
la comunidad científica y en complejos inferidos a partir de experimentos de
alto rendimiento de interacciones entre proteínas.
En este trabajo demostramos que existen diferencias
substanciales en cómo han evolucionado los complejos de proteínas entre estos
dos organismos estudiados. Un modelo previamente propuesto sobre la evolución
de los complejos de proteínas identificó complejos con núcleos centrales
formados por la interacción de proteínas homólogas. En este trabajo encontramos
evidencias que apoyan la relativa importancia de este modo de evolución en la
levadura (S. cerevisiae), pero también encontramos que este fenómeno es mucho
menos común en la evolución de los complejos procariotas (E. coli). Nuestros
resultados apuntan a profundas diferencias en el modo en el que los complejos
han evolucionado entre estos dos organismos, sugiriendo estrategias diferentes
en la evolución de los complejos
de proteínas entre procariotas y eucariotas.
3.
Predicción de nuevas proteínas
implicadas en la formación del huso mitótico en el proteoma humano.
Este trabajo ha sido desarrollado en colaboración con
distintos grupos multidisciplinares de investigadores europeos dentro de la red
de excelencia
europea en biología de sistemas ENFIN (Experimental
Network for Functional INtegration; European FP6 Programme; www.enfin.org).
Además de participar en el desarrollo de nuevas herramientas
predictivas de proteínas del huso mitótico, en este trabajo heMOS desarrollado
una plataforma integrada de predicción del huso mitótico (SPIP, Spindle
Prediction Integrated Platform; ver Figura I.1a) coordinando la integración de
las distintas series de predicciones generadas por los distintos grupos
colaboradores como son el grupo de la University College London (UCL; Prof. C. Orengo), el del Centro
Nacional de Investigaciones Oncológicas (CNIO-Madrid; Dr. Alfonso Valencia) y el
de la Technical University of Denmark (TUD; Prof. S. Brunak). La plataforma de integración y muchos de los métodos de predicción
creados en colaboración con la UCL (ver punto 1.1 y 1.2) son la base de la metodología aplicada en
este trabajo.
Las predicciones de la
plataforma SPIP fueron validadas experimentalmente por el grupo del Profesor
Erich Nigg (Max Planck Institute of Biochemistry – Munich; ver Figura
I.1b) con un porcentaje de éxito superior al 80%, cuando en protocolos
bioinformáticos anteriores se alcanzó un porcentaje máximo de éxito del 35%.
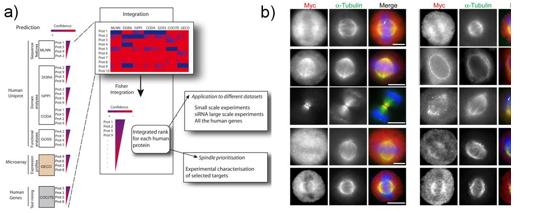
Figura I.1. A) Representación esquemática del flujo de información y tratamiento de
datos en la plataforma SPIP de integración (SPIP, Spindle Prediction Integrated Platform). B)
Imagines de algunos de los experimentos de localización en el huso mitótico de
las proteínas spindle predichas por la plataforma SPIP y validadas por el grupo
del Max-Planck en Munich.
4.
FuncNet:
Integración de métodos bioinformáticos para la predicción de alto rendimiento de
la función de proteínas.
Este trabajo está siendo
desarrollado dentro de un marco amplio de colaboración entre grupos de
investigación europea en el área de la biocomputación. La metodología,
protocolos y procesos desarrollados en los capítulos 1.1, 1.2 y 1.3 son la base
de conocimiento para la creación del servidor FuncNet. El desarrollo de FuncNet
ha sido financiado por las redes de excelencia europeas EMBRACE y ENFIN.
FuncNet es una plataforma
informática para la predicción funcional de proteínas, la cual integra
múltiples métodos predictivos bien establecidos que interaccionan con la
plataforma enviando a ésta los resultados obtenidos independientemente en cada
uno de los servidores asociados a la misma. Desde la Universidad de Málaga
hemos colaborado con el desarrollado del algoritmo y protocolo de integración
matemática de esta plataforma.
FuncNet permite a los
usuarios la integración de los resultados de diversos algoritmos predictivos en
un solo proceso de búsqueda, a la vez que ven incrementado el poder predictivo
sobre su búsqueda mediante la combinación y/o integración de los distintos
métodos (Figura I.2). Documentación técnica y los manuales de uso están
disponibles en: http://www.funcnet.eu/
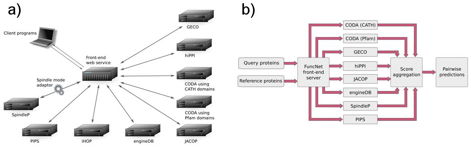
Figura I.2. A) Representación
esquemática de la plataforma FuncNet de integración y de B) su flujo y proceso
de datos.
5.
Modelado 3D del complejo de interacción
de la proteína 14-3-3 con KSR1.
Este trabajo fue desarrollado en colaboración con el grupo del Dr. José Lozano en la Universidad de Málaga. Nuestra colaboración consistió en la construcción de modelos 3D de interacción entre la proteína 14-3-3 γ y los fosfopéptidos RSKpSHE (PS297) y RTEpSVP (PS392) de la proteína KSR1 usando como modelos la estructura conocida de 14-3-3 γ unida al fosfopéptido tipo I, RAIpSLP.
El modelo resultante
(Figura I.3) mostró que todos los residuos del sitio de interacción en 14-3-3
γ estaban completamente conservados en las 7 isoformas de la proteína
14-3-3 humana. Los resultados de este modelo confirmaron que la especificidad
de la interacción está principalmente determinada por la secuencia del
fosfopéptido más que por el surco de interacción de la proteína 14-3-3. El
estudio comparativo de las distintas estructuras del complejo
14-3-3/fosfopéptido conocidas permitió determinar las relaciones entre las
variaciones de los residuos en distintas posiciones de la secuencia del
fosfopétido y los cambios conformacionales asociados en la interacción con
14-3-3 γ.
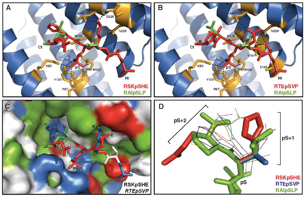
Figura I.3. Modelo 3D de la
interacción entre la proteína 14-3-3 γ y los fosfopéptidos RSKpSHE (PS297)
(a y c) y RTEpSVP (PS392) (b). Modelo 3D conformacional comparativo de 14-3-3
con distintos fosfopétidos (d).
Publicaciones:
The
functional interaction of 14-3-3 proteins with the ERK1/2 scaffold KSR1 occurs
in an isoform-specific manner.
Jagemann
LR, Pérez-Rivas LG, Ruiz EJ, Ranea JA, Sánchez-Jiménez F, Nebreda AR, Alba E,
Lozano J.
J
Biol Chem. 2008 Jun 20;283(25):17450-62. Epub 2008 Apr 21.
Predicting
protein function with hierarchical phylogenetic profiles: the Gene3D
Phylo-Tuner method applied to eukaryotic genomes.
Ranea
JA, Yeats C, Grant A, Orengo CA.
PLoS
Comput Biol. 2007 Nov;3(11):e237. Epub 2007 Oct 18.
Protein
superfamily evolution and the last universal common ancestor (LUCA).
Ranea
JA, Sillero A, Thornton JM, Orengo CA.
J
Mol Evol. 2006 Oct;63(4):513-25. Epub 2006 Oct 4.
TSEMA:
interactive prediction of protein pairings between interacting families.
Izarzugaza
JM, Juan D, Pons C, Ranea JA, Valencia A, Pazos F.
Nucleic
Acids Res. 2006 Jul 1;34(Web Server issue):W315-9.
Exploiting
protein structure data to explore the evolution of protein function and
biological complexity.
Marsden
RL, Ranea JA, Sillero A, Redfern O, Yeats C, Maibaum M, Lee D, Addou S, Reeves
GA, Dallman TJ, Orengo CA. Philos Trans R Soc Lond B Biol Sci. 2006 Mar
29;361(1467):425-40. Review.
Genome
evolution: micro(be)-economics.
Ranea
JA. Heredity. 2006 May;96(5):337-8.
Assessing
protein co-evolution in the context of the tree of life assists in the
prediction of the interactome.
Pazos
F, Ranea JA, Juan D, Sternberg MJ. J Mol Biol. 2005 Sep 30;352(4):1002-15.
Microeconomic
principles explain an optimal genome size in bacteria.
Ranea
JA, Grant A, Thornton JM, Orengo CA. Trends Genet. 2005 Jan;21(1):21-5.
Evolution
of protein superfamilies and bacterial genome size.
Ranea
JA, Buchan DW, Thornton JM, Orengo CA. J Mol Biol. 2004 Feb 27;336(4):871-87.
Heparan
sulphate mediates swine vesicular disease virus attachment to the host cell.
Escribano-Romero
E, Jimenez-Clavero MA, Gomes P, García-Ranea JA, Ley V.
J
Gen Virol. 2004 Mar;85(Pt 3):653-63.
The
small GTP-binding protein, Rhes, regulates signal transduction from G
protein-coupled receptors.
Vargiu
P, De Abajo R, Garcia-Ranea JA, Valencia A, Santisteban P, Crespo P, Bernal J.
Oncogene.
2004 Jan 15;23(2):559-68.
p23
and HSP20/alpha-crystallin proteins define a conserved sequence domain present
in other eukaryotic protein families.
Garcia-Ranea
JA, Mirey G, Camonis J, Valencia A.
FEBS
Lett. 2002 Oct 9;529(2-3):162-7.
Mapping
of linear epitopes on the capsid proteins of swine vesicular disease virus
using monoclonal antibodies.
Borrego
B, García-Ranea JA, Douglas A, Brocchi E.
J
Gen Virol. 2002 Jun;83(Pt 6):1387-95.
Characterization
of neutralization sites on the circulating variant of swine vesicular disease
virus (SVDV): a new site is shared by SVDV and the related coxsackie B5 virus.
Borrego
B, Carra E, García-Ranea JA, Brocchi E.
J
Gen Virol. 2002 Jan;83(Pt 1):35-44.
Immune
recognition of swine vesicular disease virus structural proteins: novel
antigenic regions that are not exposed in the capsid.
Jiménez-Clavero
MA, Douglas A, Lavery T, Garcia-Ranea JA, Ley V.
Virology.
2000 Apr 25;270(1):76-83.
Model
of the ran-RCC1 interaction using biochemical and docking experiments.
Azuma
Y, Renault L, García-Ranea JA, Valencia A, Nishimoto T, Wittinghofer A.
J
Mol Biol. 1999 Jun 18;289(4):1119-30.
Effector
recognition by the small GTP-binding proteins Ras and Ral.
Bauer
B, Mirey G, Vetter IR, García-Ranea JA, Valencia A, Wittinghofer A, Camonis JH,
Cool RH.
J
Biol Chem. 1999 Jun 18;274(25):17763-70.
RhoGAPs
and RhoGDIs, (His)stories of two families.
Zalcman
G, Dorseuil O, Garcia-Ranea JA, Gacon G, Camonis J.
Prog
Mol Subcell Biol. 1999;22:85-113. Review. No abstract available.
Distribution
and functional diversification of the ras superfamily in Saccharomyces
cerevisiae.
Garcia-Ranea
JA, Valencia A.
FEBS
Lett. 1998 Sep 4;434(3):219-25.
Search
for ancient patterns in protein sequences.
Thode
G, García-Ranea JA, Jimenez J.
J
Mol Evol. 1996 Feb;42(2):224-33.