

Función y redes de proteínas
ACTIVIDAD INVESTIGADORA
Predicción, modelado y análisis topológico de redes de proteínas para los proteomas completos de H. sapiens y Saccharomyces cerevisiae.
En estrecha colaboración con el grupo de la profesora Christine Orengo en la University College London (UCL), hemos modelado dos tipos de redes de interacción entre proteínas para los proteomas completos de H. sapiens y S. cerevisiae, redes que denominamos KnowledgeGram (KG) y PredictoGram (PG) por el origen de los datos usados en su construcción. Las redes KG integran la información sobre asociación funcional de pares de proteínas obtenida de bases de datos de “conocimiento” biológico y/o experimental, tales como: HRPD, MINT, Intact, Reactome, Kegg, Gene Ontology (GO) y FunCat. Mientras que las redes PG, integran mediante el método Fisher, predicciones ab-initio de interacción entre proteínas obtenidas por tres métodos bioinformáticos distintos diseñados y desarrollados en éste mismo trabajo, como son: GECO (Gene Expressison COrrelation). CODA (Co-Ocurrence Domain Analysis); y hiPPI (homology inherited Protein-Protein Interactions).
En
este trabajo demostramos que las redes PG, basadas en predicciones,
presentan las mismas características topológicas de las
redes biológicas experimentales KG. También demostramos
que las redes PG contienen información funcional clave en el
modelado de los sistemas biológicos que componen estos dos
organismos estudiados, información que en un alto porcentaje
(80%-90%) no está recogida en las redes KG basadas en
evidencias experimentales.
Entrevista en Biotechniques:
Recientemente nuestro trabajo Finding the
"Dark Matter" in Human and Yeast Protein Network Prediction and Modelling ha suscitado el interés de la revista Biotechniques.com, Journal internacional de métodos en ciencias de la vida ubicado en Nueva York; a raíz del cuál surge una entrevista en la que se describe el trabajo de nuestro grupo en esta linea de investigación de una forma altamente divulgativa y sin pérdida de rigurosidad.
Biotechniques. com y la "Dark Matter"
Modelado y análisis evolutivo de los complejos de proteínas en los proteomas completos de Escherichia coli y Saccharomyces cerevisiae.
En colaboración con la UCL hemos modelado con alta precisión los complejos de proteínas para los proteomas completos de E. coli y S. cerevisiae, basándonos en complejos de proteínas comúnmente aceptados por la comunidad científica y en complejos inferidos a partir de experimentos de alto rendimiento de interacciones entre proteínas.
En este trabajo demostramos que existen diferencias substanciales en cómo han evolucionado los complejos de proteínas entre estos dos organismos estudiados. Un modelo previamente propuesto sobre la evolución de los complejos de proteínas identificó complejos con núcleos centrales formados por la interacción de proteínas homólogas. En este trabajo encontramos evidencias que apoyan la relativa importancia de este modo de evolución en la levadura (S. cerevisiae), pero también encontramos que este fenómeno es mucho menos común en la evolución de los complejos procariotas (E. coli). Nuestros resultados apuntan a profundas diferencias en el modo en el que los complejos han evolucionado entre estos dos organismos, sugiriendo estrategias diferentes en la evolución de los complejos de proteínas entre procariotas y eucariotas.
Predicción de nuevas proteínas implicadas en la formación del huso mitótico en el proteoma humano.
Este trabajo ha sido desarrollado en colaboración con distintos grupos multidisciplinares de investigadores europeos dentro de la red de excelencia europea en biología de sistemas ENFIN (Experimental Network for Functional INtegration; European FP6 Programme; www.enfin.org).
Además de participar en el desarrollo de nuevas herramientas predictivas de proteínas del huso mitótico, en este trabajo heMOS desarrollado una plataforma integrada de predicción del huso mitótico (SPIP, Spindle Prediction Integrated Platform; ver Figura I.1a) coordinando la integración de las distintas series de predicciones generadas por los distintos grupos colaboradores como son el grupo de la University College London (UCL; Prof. C. Orengo), el del Centro Nacional de Investigaciones Oncológicas (CNIO-Madrid; Dr. Alfonso Valencia) y el de la Technical University of Denmark (TUD; Prof. S. Brunak). La plataforma de integración y muchos de los métodos de predicción creados en colaboración con la UCL (ver punto 1.1 y 1.2) son la base de la metodología aplicada en este trabajo.
Las predicciones de la plataforma SPIP fueron validadas experimentalmente por el grupo del Profesor Erich Nigg (Max Planck Institute of Biochemistry – Munich; ver Figura I.1b) con un porcentaje de éxito superior al 80%, cuando en protocolos bioinformáticos anteriores se alcanzó un porcentaje máximo de éxito del 35%.
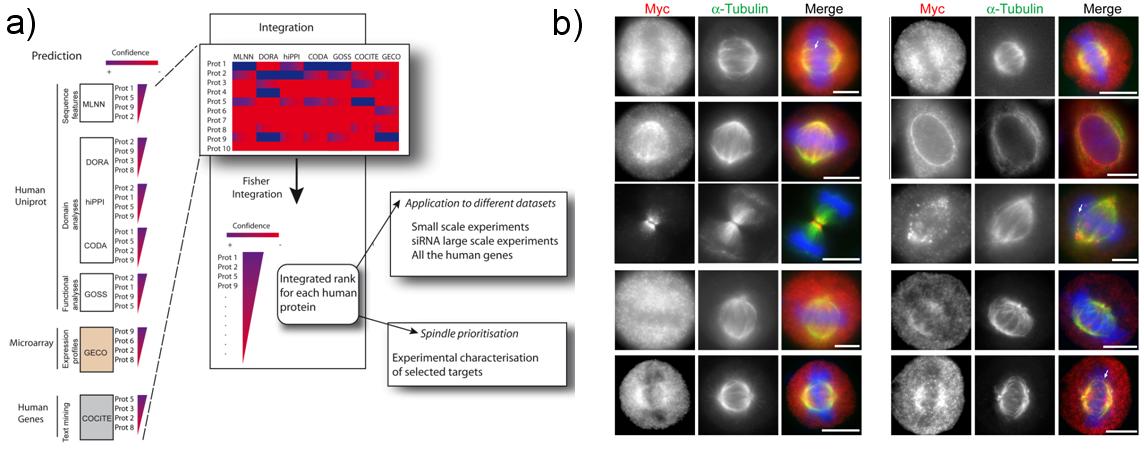
Figura I.1. A) Representación esquemática del flujo de información y tratamiento de datos en la plataforma SPIP de integración (SPIP, Spindle Prediction Integrated Platform). B) Imagines de algunos de los experimentos de localización en el huso mitótico de las proteínas spindle predichas por la plataforma SPIP y validadas por el grupo del Max-Planck en Munich.
FuncNet: Integración de métodos bioinformáticos para la predicción de alto rendimiento de la función de proteínas.
Este trabajo está siendo desarrollado dentro de un marco amplio de colaboración entre grupos de investigación europea en el área de la biocomputación. La metodología, protocolos y procesos desarrollados en los capítulos 1.1, 1.2 y 1.3 son la base de conocimiento para la creación del servidor FuncNet. El desarrollo de FuncNet ha sido financiado por las redes de excelencia europeas EMBRACE y ENFIN.
FuncNet es una plataforma informática para la predicción funcional de proteínas, la cual integra múltiples métodos predictivos bien establecidos que interaccionan con la plataforma enviando a ésta los resultados obtenidos independientemente en cada uno de los servidores asociados a la misma. Desde la Universidad de Málaga hemos colaborado con el desarrollado del algoritmo y protocolo de integración matemática de esta plataforma.
FuncNet permite a los usuarios la integración de los resultados de diversos algoritmos predictivos en un solo proceso de búsqueda, a la vez que ven incrementado el poder predictivo sobre su búsqueda mediante la combinación y/o integración de los distintos métodos (Figura I.2). Documentación técnica y los manuales de uso están disponibles en: http://www.funcnet.eu/
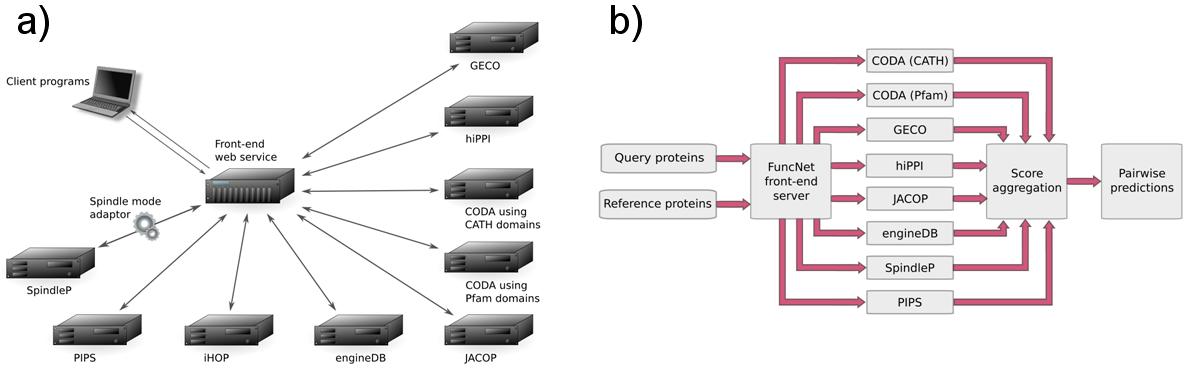
Figura I.2. A) Representación esquemática de la plataforma FuncNet de integración y de B) su flujo y proceso de datos.
Modelado 3D del complejo de interacción de la proteína 14-3-3 con KSR1.
Este trabajo fue desarrollado en colaboración con el grupo del Dr. José Lozano en la Universidad de Málaga. Nuestra colaboración consistió en la construcción de modelos 3D de interacción entre la proteína 14-3-3 γ y los fosfopéptidos RSKpSHE (PS297) y RTEpSVP (PS392) de la proteína KSR1 usando como modelos la estructura conocida de 14-3-3 γ unida al fosfopéptido tipo I, RAIpSLP.
El modelo resultante (Figura I.3) mostró que todos los residuos del sitio de interacción en 14-3-3 γ estaban completamente conservados en las 7 isoformas de la proteína 14-3-3 humana. Los resultados de este modelo confirmaron que la especificidad de la interacción está principalmente determinada por la secuencia del fosfopéptido más que por el surco de interacción de la proteína 14-3-3. El estudio comparativo de las distintas estructuras del complejo 14-3-3/fosfopéptido conocidas permitió determinar las relaciones entre las variaciones de los residuos en distintas posiciones de la secuencia del fosfopétido y los cambios conformacionales asociados en la interacción con 14-3-3 γ.
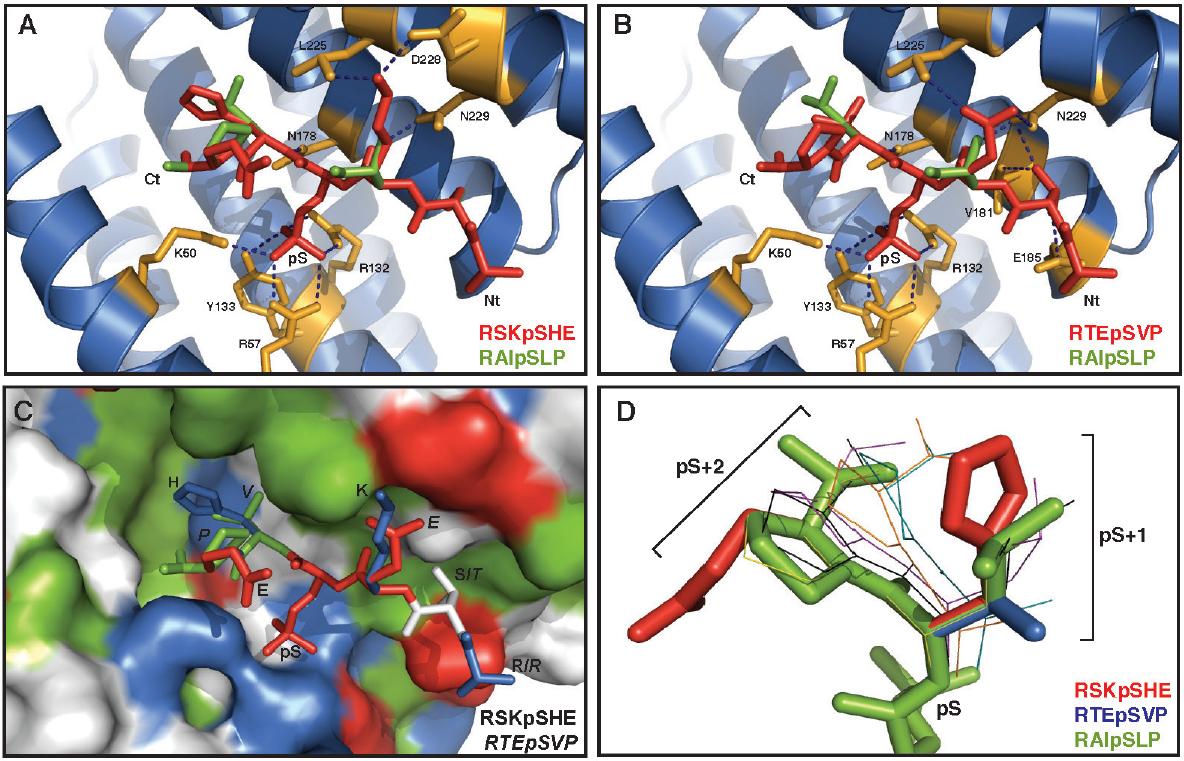
Figura I.3. Modelo 3D de la interacción entre la proteína 14-3-3 γ y los fosfopéptidos RSKpSHE (PS297) (a y c) y RTEpSVP (PS392) (b). Modelo 3D conformacional comparativo de 14-3-3 con distintos fosfopétidos (d).
Sistemas de Microscopía.
Es la más reciente linea de investigación de nuestro grupo. Varios laboratorios europeos punteros en Biología computacional entre el que se encuentra el nuestro han creado una red europea de excelencia denominada "Systems Microscopy NoE" y financiada por la comisión europea de investigación dentro del marco EU-FP7.
Esta red nace con la idea de automatizar complejos procesos de análisis de genotipado y posterior reconocimiento del fenotipo resultante mediante tratamiento de imágenes de microscopía. Con este próposito aunan sus esfuerzos los laboratorios de investigadores europeos tales como Jan Ellenberg, Daniel Gerlich o Wolfgang Huber del EMBL de Alemania, Uri Alon y Benny Geiger del instituto Weizmann en Israel, Olli Kallioniemi de la Universidad de Helsinki en Finlandia o Jason Swedlow de la Universidad de Dundee en Escocia.

Figura I.4. Cartel en el que se publicita la puesta en marcha de la red mediante un simposium que acogerá la Universidad de Málaga el próximo 21 de Febrero de 2011. Como ponentes invitados se encuentran Steven Altschuler y Lani Wu de la Universidad de Texas en EEUU o Rick Horwitz de la Universidad de Virgina también en EEUU.
ENLACES DE INTERÉS Y COLABORACIONES:
CIBER-ER - KHAOS - UCL - COST - CSIC - REMA - ICIII - MICINN - UMA - PAI - FUNCNET - ENFIN - Univ.Utah - Matlab - Biotechniques.com
Diseñado por Proyecta Científica
